the misfolded nightmare: prions
welcome to A Teetering Vulture! a newsletter about various science stuff as well as the life happenings of its author, Taylor.

In my opinion, few medical topics are scarier than the one I’m about to talk about. As someone who has contended with not insubstantial levels of health anxiety at various points in her life, it is somewhat astounding that I’ve been able to research this topic and talk to people about it without ending the day wide awake in bed, terrified of my own body and its stochastic machinations, and on the cusp on a panic attack. It’s a testament to the progress I’ve made over the years in responding to my health anxiety, I suppose. Or it’s just that it’s easier to prevent myself from getting stuck in a cycle of fearful rumination when my life is somewhat stable, which it is, currently. I’m fortunate enough that nothing is going too terribly wrong, at the moment (Knock on wood). Woohoo! So let’s talk about prions, then.
(Before I begin, however, I would like to address readers of this who have dealt, or are dealing, with health anxiety perhaps in similar ways that I have. If you know you are prone to hearing about a medical issue and becoming concerned about developing or having that medical issue, I kindly offer caution going forward. Prions are no joke, and I fear they have the capacity to send anyone into an anxious or existential spiral. So this is your warning. Feel free to take a moment to make a decision about reading any further.)
For those still with me, I’d like you to take a moment, grab a small piece of paper or open a note on your phone, your computer. Or simply prepare to make a short mental list in your head. Then, take a minute or two to record all the causes of disease that you can think of. In humans, in animals, in trees—in whatever living thing, as long as it causes an illness of some kind. Cause is the key word here. I’m looking for categories, not specific illnesses. For instance, “the flu” is not a cause of illness, but an illness itself.
Once you’ve finished, we can carry on together.
A disease may be caused by a virus, one like SARS-CoV-2, a little packet of genetic information and perhaps a few other biomolecules designed to co-opt a host’s cellular machinery to propogate itself. Or it may be a bacteria, some nefarious single-celled organism like E. coli, which can duplicate itself rapidly inside your digestive tract and release an intestine-damaging toxin. Or possibly a fungus—or an amoeba—or a protozoan parasite like Plasmodium, the causal agent of Malaria—or even a parasitic worm. An aberrated gene in a cell, too—one that allows a cell to prioritize its life and reproduction over the lives of cells that are essential for the livelihood of the organism in which it inhabits, which is cancer. There are many other genetic mutations and chromosomal mutations that can cause disease, as well.
A prion does not necessarily fit into any of these categories, which makes it, to put it simply, rare and very weird. It can sort of fit into one of these categories, and I’ll discuss how soon, but basically, it’s astoundingly unique and strange. This uniqueness is definitely part of the reason it is so scary, because it means we haven’t been considering ways to stop it for nearly as long (or on the same scale) as we have been considering ways to stop other disease-causing menaces.
A prion is a protein. That is the type of molecule that it is: a polymer comprised of linkages between twenty-three types of amino acids. What differentiates it from the numerous non-disease causing proteins in a body is it’s shape, and specifically the way that is folded. A prion is in essence a protein that has become improperly folded—rearranged abnormally at the cellular level. It’s misfolded nature allows it to catalyze the misfolding of other proteins. A rapid accumulation of misfolded proteins leads to disease presentation, as this cascade has all sorts of disastrous consequences at molecular and physiological levels. In all mammals, there is only one protein that can become a prion, and it’s the PrP protein, also called (in ominous reference to its potential future form) the prion protein.
In its normal, non-prion state, the PrP protein has been found to play a non-essential role in neuron maintenance in the brain. For a long time, scientists struggled to determine the native function of PrP, but studies in which scientists have removed PrP from the brains of mice have given us clues as to what it does. Scientists have also identified people who naturally lack PrP because of a mutation in the gene that encodes the protein. These people are all apparently healthy.
The etiology of prion disease has been called “sporadic,” for its rare incidence and its often unknown underlying cause. Creutzfeldt-Jakob disease is the name given to prion disease that develops in humans seemingly at random, when it is a mystery what has induced the PrP protein to become a prion.
However, there are certain germline mutations in the PrP-encoding gene that can make some people much more predisposed to prion disease. About 5-10% of prion disease cases are a result of a hereditary origin. And a smaller percentage still acquire prion disease from coming into contact with infected tissues. This can occur through ingestion or nosocomial means.
Horrifically, as of 2024, the outcome for anyone who acquires prion disease is death. There is no cure for prion disease, and it is resoundingly fatal. Prion disease is neurodegenerative, the brain incapable of overcoming the damage prions and prion aggregates cause to its spongiform and neuronal tissues once they’ve been introduced to them. Symptoms of prion disease in humans are diverse, ranging from increasingly severe dementia to insomnia, paranoia, anxiety, depression, and loss of motor control. The central challenge of eradicating prions from the brain is difficult both because of a need for therapeutics that can cross the blood-brain barrier, and because of a prion’s chemical structure, which just so happens to make it nigh on invincible and immortal. Outside of a body, a prion is immune to degradation via heat, it is incapable of being digested proteolytically, and it is insoluble, meaning that chemical detergents have no effect. Consider that surgical instruments sterilized and left unused for years have been found to still harbor prions. It’s a pathogenic foe that seems almost impossible to contend with. And inside of a brain, incapacitating something so imperishable presents a horrendously intimidating task for scientists.
While rare in humans, with case numbers of around 1-2 people per million every year, prions themselves aren’t all that rare. The most common place a person might encounter prion disease is, unsettlingly, right in their backyard. Chronic wasting disease (CWD) is a transmissible prion disease of deer and other cervid species (elk, moose, etc.) that has been of increasing concern in North America since the early 1980s, when it was first discovered in a wild animal. The disease is, very fortunately, contained to cervids, though scientists have been keeping a wary eye on its spread throughout the United States and elsewhere. A group of public health experts arranged by the Center for Infectious Disease Research and Policy (CIDRAP) is assessing the potential for the disease to make the jump from cervids into humans, a spillover event that could prove apocalyptic, and to formulate a response plan. So far, such an event has been deemed unlikely. Though potentially not impossible.
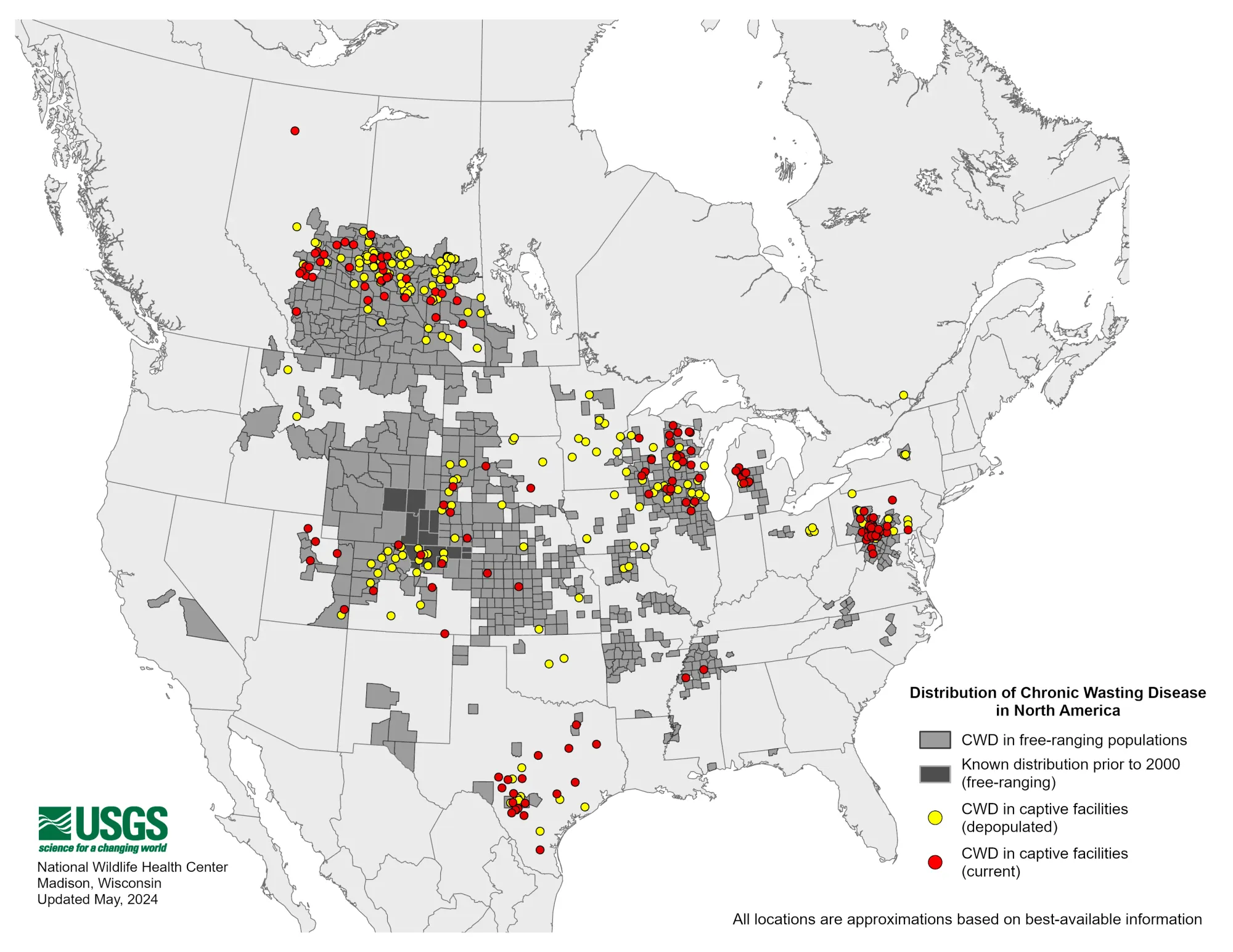
The presentation of CWD is similar in deer as it is in humans. Because prions eat away at spongiform brain tissue, deer lose mental and motor functions over time. They often become nervous, confused, stumbling—especially in the final days before they die. Sometimes the disease takes years to manifest or to weaken a deer to a point where it can no longer function well enough to survive. The same is true in humans: prion disease can have a long latency period in which no symptoms present themselves. This is another facet of the disease which makes it difficult to contend with, as it is hard to track the cause or origin of a disease that has been sitting quietly in the brain of a person for years.
CWD is steadily spreading around the United States. Late in 2023, Yellowstone National Park recorded its first cases. And in May of 2024, California became the most recent of 32 other states to have reported deer with CWD.
In order to prevent human exposure to CWD, which could increase the likelihood of the disease morphing into a form that can infect humans, scientists are working to develop a diagnostic method that will make it possible for deer to be tested for CWD rapidly in the field. If such technology is developed, those that come in regular close contact with deer, such as hunters, would have the tools needed to determine if a deer is better left untouched, or if it’s safe for handling or consumption. Consumption of venison in the United States is not uncommon, and considering the overpopulation of deer in many regions of the country, hunting is often beneficial to communities of native plants and the species that rely on those plants. But ingestion of CWD-infected deer meat puts humans in extremely close contact with prions. While those prions remain primarily localized in the brain and lymph system of deer, studies have found that prions also end up in the blood of deer, in addition to feces and urine. Meaning its not unlikely they are also in the tissues people are eating.
As of now, diagnostic methods like immunohistochemistry (IHC), enzyme-linked immunosorbent assays (ELISA), and Western blots are slow and require a laboratory. More rapid methodology like real time quacking-induced conversion (RT-QuIC), which uses blood to diagnose CWD antemortem or postmortem, are in development but still requires equipment that is not amenable to field use. If a rapid diagnostic test does become available, it may also allow scientists to test more efficiently for environmental prevalence of prions. If deer are shedding prions in their waste, then the spaces they occupy are presumably full of prions, and interminably so, because of how long prions are inclined to stick around in the world.
Prion endurance has sinister implications. If once created, prions persist, then unlike a certain strain of virus or bacteria, there is no guarantee of eventual eradication of prions. More prions existing in deer, humans, and other organisms means more prions in the world, sort of forever. Continually increasing numbers of prions will result in ever more opportunities for them to affect us more severely than they do now. Considering a future where prion disease is not rare, but still always fatal, is frankly terrifying.
So, now knowing all of this, I would imagine it’s reasonable to be asking if all hope is lost. To be asking: is this it, the pathogen that will bring us all down one day?
To answer both those questions: hopefully, no, it’s not. Unsurprisingly, there are a myriad of very highly skilled people who are paying very close attention to prions, and those people are also working very hard to make sure that this becomes a surmountable disease one day. Some of the people at the forefront of prion disease research are moreover deeply and personally invested in a cure. Such investment may hopefully accelerate the development of a cure, ensuring it comes sooner rather than later.
Dr. Sonia Vallabh and her husband Dr. Eric Vallabh Minikel direct a human prion disease research team at the Broad Institute at the Massachusetts Institute of Technology (MIT). Dr. Sonia Vallabh is one of the rare humans who has a mutation in her PrP gene that makes it highly likely she will one day contract prion disease. After her mother passed away from the disease suddenly and unexpectedly in 2012, she and her husband made abrupt career changes, pivoting from their prior, unscientific jobs into biomedical research. Dr. Minikel has kept a blog since the beginning of this massive life shift, which he also describes as like one half of his brain, a place where he stores notes and keeps track of advancements in knowledge about prion disease and how to combat it. It now has over a decade’s worth of entries dedicated to understanding prions. Their team is interested both in drugs that may have antiprion activity or that may combat symptoms, and also in biochemical techniques that may allow for the removal of PrP from the brain altogether, thereby rendering individuals nearly invincible against prion disease; without PrP to serve as seeds for new prions, a person cannot be affected much even if minimal amounts of prions did make their way into the brain.
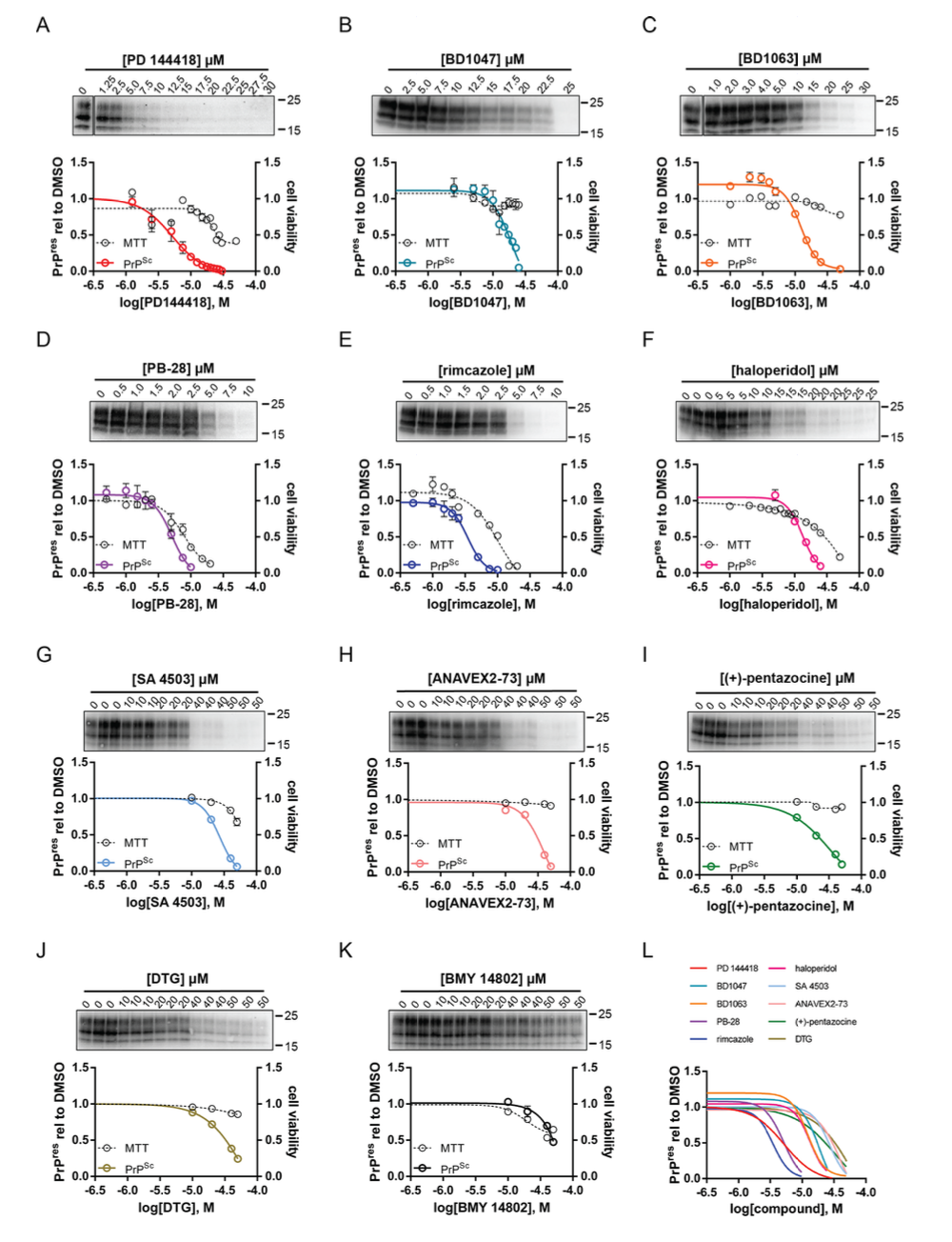
Other researches are working on discovering antiprion compounds that could be used to combat the neurodegenerative activity of prions. One team, from a paper published in 2024, stumbled across ten particularly potent antiprion compounds while researching other antiprion drugs. These compounds reduce synaptotoxicity of prions, meaning they reduce the ability of a prion to degrade the dendrites of neural synapses. It’s possible some of these compounds have other mechanisms of action as well. Questions remain about whether or not they may interact with prions directly to stymie their deleterious actions, or work elsewhere in a manner that, downstream, results in disruption of prion activity. Regardless, the discovery of more and more antiprion compounds, some of which have only come to light because of adjacent scientific work being done to study COVID-19, is promising. There are those who are becoming optimistic that prion disease won’t, in the near future, be as intimidating and harrowing as it is now.
In fact, in a recent blog post, Dr. Minikel wrote that right now feels like “the end of the beginning” of prion disease research. Humanity is finally getting a firmer grasp on this disease—there is burgeoning hope—and it’s in large part thanks to people who are looking this scary disease in the eye every day, for the sake of us all.
Information for the article was obtained from:
RT-QuIC detection of chronic wasting disease prion in platelet samples of white-tailed deer ✼ Prions are forever ✼ As ‘Zombie’ Deer Disease Spreads, Scientists Look for Answers ✼ Sigma Receptor Ligands Are Potent Antiprion Compounds that Act Independently of Sigma Receptor Binding ✼ Ohio Department of Natural Resources ✼ Erik Vallabh Minikel's Blog